|
Research programs Research
Interests Techniques Representative Projects FAQ
Representative projects in cancer therapy:
- Development of water-soluble highly-potent
albumin-binding drugs
Novel
thiol-binding hydrazide-containing linkers have been developed to create
drug-carrier systems that allow effective
and controlled release of highly-potent drugs specifically at the acidic tumor site. Once bound to a carbonyl-containing drug, the
obtained acid-sensitive hydrazone bond has an improved stability under both physiological and acidic
conditions. The linkers have proven to be fruitful to
create drug carrier systems not only for conventional chemotherapeutics but
also for highly potent drugs demonstrating superior antitumor efficacy versus
the free drugs in mice experiments.
Initially, a library of aromatic hydrazides
bearing maleimide has been synthesized. The
aromatic linker was substituted with electron withdrawing groups
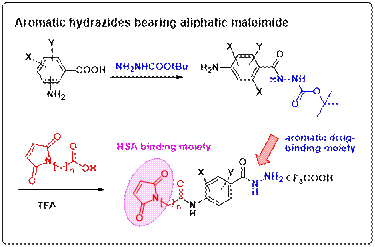 
Following
the synthesis, the next step was to study the effect of the substituents on
the drug release and hence the pharmacokinetic profile of the drug. For
this purpose, 1) hydrazone derivatives have been
synthesized with nemorubicin, a
carbonyl-containing anthracycline as a model drug, 2) and the human serum
albumin (HSA) conjugates of these hydrazones were
then prepared in vitro with
commercial HSA. Stabilities of hydrazones in
aqueous media at the physiological/injection conditions, and the drug
release profiles of the HSA conjugates under acidic conditions were
studied and analyzed by HPLC and LC-MS.
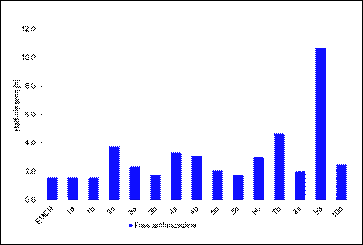
The
results show that 1) the aliphatic maleimides were remarkably more stable
than the aromatic maleimides at the physiological conditions, 2) higher
stability, and thus slower drug release from the conjugate, was obtained for the hydrazones substituted at their ortho-position. Because many clinically established
chemotherapeutic agents are water-insoluble, the electron withdrawing
sulfonate group was preferred to develop sulfonate-based water-solubilizing
linkers.
Drugs
lacking carbonyl groups have been derivatized, and the toxicity of the new
derivatives was studied both in vitro and in vivo. Such
carbonyl-containing compounds have been used together with the lead linker
below to develop novel highly-potent auristatin E- and maytansinoid-based
albumin-binding prodrugs with superior anticancer efficacy in several human
cancer xenograft models in nude mice:

- Development of imaging agent for determining albumin uptake in tumors
SPECT/CT imaging is a preclinical and clinical technique that is
used to determine the biodistribution and tumor uptake of radiolabeled peptides and macromolecules. The purpose of
this project was to develop an albumin-binding compound as an imaging agent for the detection of
endogenous albumin and its tumor accumulation in human tumor xenograft models.
Structural
design and syntheses: The general chemical structure of the
precursors for the imaging agent
was designed to contain i) a maleimide group as an albumin-binding moiety
that binds rapidly and specifically to the amino acid
cysteine-34 of circulating albumin, and ii) the acyclic chelating agent diethylenetriaminepentaacetic
acid (DTPA),
which is capable of binding the gamma emitter indium-111 (111In, half-life of 2.8
days) suitable for SPECT imaging.
Initially, four compounds differing in the
length of the maleimide chain were synthesized and purified by HPLC. In
order to select one compound for the radiolabeling and imaging studies, the
compounds were initially reacted with non-radioactive indium(III)chloride
and subsequently bound to the cysteine-34 position of human serum in
analogy to our experience with albumin-binding drugs. HPLC analyses of the resulting data showed
comparable results for all compounds for relevant parameters.
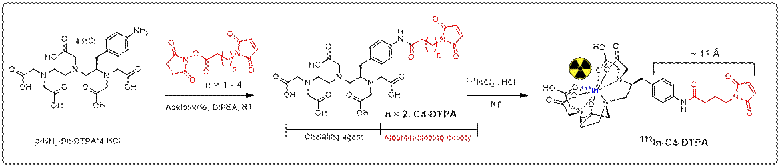
We chose C4-DTPA for further development because the ~12 Å maleimide chain length of the molecule is
in good agreement with the accessible 10-12 Å deep cavity of the
cysteine-34 position of albumin.
Radiochemical
labeling and albumin-binding
properties of the imaging agent111In-C4-DTPA: The albumin-binding imaging agent 111In-C4-DTPA
was custom-synthesized and reproducibly produced with high radiochemical purity > 99 % from
radiolabeling its
precursor C4-DTPA with 111InCl3 in HCl solution. The albumin-binding properties of 111In-C4-DTPA
were studied in vitro in both
human and murine serum. A shown by radio-HPLC, the imaging agent was bound rapidly to albumin, and the resulting
radiolabeled albumin conjugates showed high stability in murine and human
serum.
In vivo SPECT/CT
imaging of tumor-bearing mice: For
evaluating the biodistribution and tumor uptake of the imaging agent, single-photon emission computed tomography/X-ray-computed tomography
(SPECT/CT) imaging studies were carried out (contract
research) in different human tumor xenograft models. After intravenous
administration of 111In-C4-DTPA,
the tumor bearing mice were
imaged by SPECT/CT over 72 h at 4-6 time
points.
|
Distinct radioactivity was observed in
the growing tumors as can be discerned from representative 2D and 3D
SPECT/CT images with a maximum measured in the tumor at ~ 24 h
post-injection.
Results from
these experiments show that the imaging agent 111In-C4-DTPA binds rapidly and
selectively to circulating albumin after intravenous administration and
demonstrates a clear accumulation in the tumor substantiating the drug
delivery approach of binding low-molecular weight drugs to the
cysteine-34 position of albumin as the drug carrier.
Such an imaging agent has the
potential for use in personalized medicine as a companion diagnostic for
identifying patients for targeted therapy with albumin-binding drugs.
|
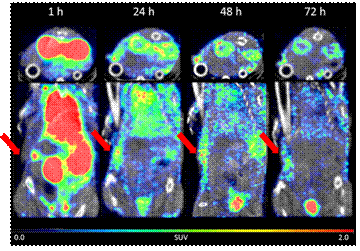
Representative images of a mouse of
the ovarian cancer OVXF899 tumor-bearing mice presented as transverse
plane (top) and dorsal plane (bottom).
|
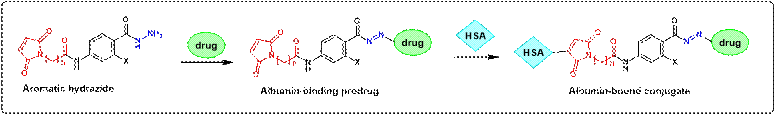
- Development of dual-acting prodrugs as the next
generation of albumin-binding prodrugs
Albumin
(HSA) is used as a drug carrier due to its uptake in the tumors. In situ coupling to albumin is achieved
by intravenous application of a maleimide bearing prodrug that binds
rapidly and selectively to the cysteine-34 position of
circulating albumin. This
targeting approach is based on the release of the albumin-bound drugs
predominantly at the tumor site due to the
incorporation of a cleavable bond between the drug and the carrier. It is a
clinically validated technology of increasing the therapeutic index of
anticancer agents.
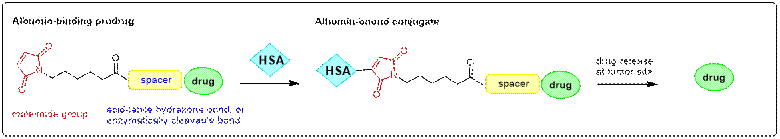
In order
to improve the therapeutic efficacy of albumin-binding prodrugs, we have
developed new platform technologies based on dual-acting prodrugs as the
next generation of albumin-binding prodrugs.
|
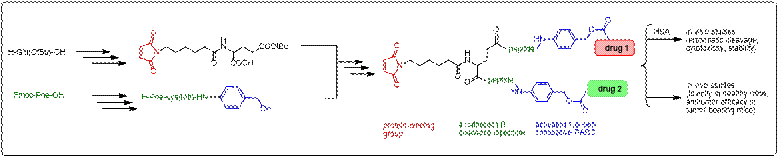
I designed and performed the
synthesis of the first examples of such compounds. In a “combination
chemotherapy” approach, two drugs were introduced in the prodrug. In a “circumventing
multidrug resistance (MDR)” approach, one drug and one MDR-inhibitor were
integrated in the prodrug.
|

|
The
synthesis is based on a protein-binding bifunctional linker that can be
synthesized from commercially available amino acids. Preferred spacers
are enzymatically cleavable peptides linked to self-immolative
para-aminobenzyloxycarbonyl (PABC) spacer.
Controlled
release of the drugs can be varied by using peptides with different
enzymatic cleavage rates. The new prodrugs were isolated by preparative
HPLC and they underwent an in vitro and in vivo evaluation.
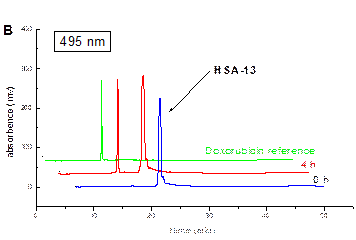
The synthesis pathway was fruitfully applied in other projects.
Below is a
representative example for HPLC drug release study of the HSA conjugates.
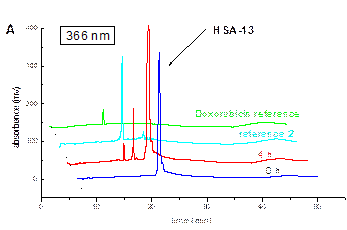 
|